
실무 관점 분석
– 허가 준비, 제조소 총람, 공급망까지 확대되는 책임 범위 –
"개편된 구조에서는 문서 품질보다
'현장 일치성' 확보가 훨씬 중요해졌으며,
시스템 기반 점검이 요구됩니다."
– 식약처 의약품 정책설명회 발표자료 (2025)
🔹 서론 – 문서보다 중요한 것은 '운영 일치성'입니다
- 제도 개편의 핵심 변화:
- 이번 제도 개편에서 실무자가 주목해야 할 가장 중요한 변화는
'허가와 GMP 평가의 병렬 진행'이 아닌, 현장 운영 상태와 평가 자료의 일치성입니다. - 형식적인 문서 제출보다는 운영 실태 기반의 설명과
위험 기반 평가 대응을 위한 시스템 정비가 핵심입니다.
- 이번 제도 개편에서 실무자가 주목해야 할 가장 중요한 변화는
- 확대되는 대응 방식:
- 사전상담, 종합보고서, 공급자 평가는 서로 긴밀히 상호 연결되어 있으며,
대응 방식도 개별 문서 작성에서 조직 전체 관리 중심으로 확장되고 있습니다.
- 사전상담, 종합보고서, 공급자 평가는 서로 긴밀히 상호 연결되어 있으며,
1️⃣ 제조소 총람 – GMP 종합보고서로의 진화
- 기존 GMP 자료:
- 기존에는 조직도, 제조공정, 밸리데이션, 기록서 양식 등
총 11종의 자료를 각각 개별적으로 제출했습니다.
- 기존에는 조직도, 제조공정, 밸리데이션, 기록서 양식 등
- 개편된 'GMP 종합보고서':
- 이번 개편으로 기존 자료들을 다음 4개 항목으로 통합하여
'GMP 종합보고서'로 일원화합니다.- ① 일반현황 및 인프라
- ② 품질보증체계
- ③ 문서관리 및 기록
- ④ 시설·설비 관리
- 이번 개편으로 기존 자료들을 다음 4개 항목으로 통합하여
- 종합보고서의 핵심:
- 종합보고서는 단순한 통합 문서가 아닙니다. 심사자가
단일 문서만으로 제조소의 전반적인 품질책임체계를 파악할 수 있도록 구성해야 합니다. - 문서 간 논리적 연결성과 현장 일치성이 핵심입니다.
- 종합보고서는 단순한 통합 문서가 아닙니다. 심사자가
🔸 기존 11종 자료 목록 vs 4종 통합 구조 비교표
🔸 GMP 종합보고서 항목별 상세 구성도 및 작성 흐름 예시

✅ 실무 대응 체크
- 총람은 추상적 설명이 아닌 실제 운영 방식에 기반한 구체적 사례로 구성해야 합니다.
- 조직도, 공정도, 품질시스템 절차는 상호 정합성을 유지하며,
심사자가 명확하게 문서의 흐름을 파악할 수 있어야 합니다. - 공정 흐름도, 시스템 구성도, 대표 배치기록 양식 등 시각자료를
활용하여 설명력을 높이는 것이 좋습니다.
2️⃣ 사전상담과 병렬심사 대비 사전점검 체계
- 병렬심사 구조로의 전환:
- GIFT 제도 도입으로 허가 절차가 GMP 평가, CMC 심사,
임상자료 검토를 병렬적으로 수행하는 구조로 전환되었습니다.
- GIFT 제도 도입으로 허가 절차가 GMP 평가, CMC 심사,
- 사전상담의 중요성:
- 이에 따라 허가 신청 이전 사전상담 단계에서 심사 전략을 구체화하고,
자료 간 정합성을 사전에 확보하는 것이 필수가 되었습니다.
- 이에 따라 허가 신청 이전 사전상담 단계에서 심사 전략을 구체화하고,
- 불일치 시 발생 가능한 문제점:
- 특히 사전상담의 전략, 실제 제출 자료, GMP 종합보고서의
제조소 운영 내용이 서로 일치하지 않을 경우,
병렬심사 전체가 지연되거나 보완이 요구될 수 있습니다.
- 특히 사전상담의 전략, 실제 제출 자료, GMP 종합보고서의
🔸 병렬심사 흐름도 내 사전상담-문서연계 흐름도
🔸 허가 신청 전 체크리스트 예시
| 점검 항목 | 주요 문서/자료 | 확인 포인트 |
| 사전상담 전략 정합성 확인 |
- 사전상담 요약서 - 제품 개발 이력서 |
✔️ 병렬 적용 전략 수립 여부 ✔️ 허가총괄과 피드백 반영 여부 |
| CMC 자료 구성 상태 점검 |
- CTD 3.2.P 작성본 - 밸리데이션 개요 - 시험 항목 설정서 |
✔️ CQA 정의 및 설정 근거 명확성 ✔️ 시험법 유효성 확보 여부 |
| 제조소 운영 상태 점검 |
- 제조소 조직도 - 공정 흐름도 - QA 자체점검 보고서 |
✔️ 실제 공정 흐름과 제출자료 일치 여부 ✔️ 현장 운영 상태 확인 완료 여부 |
| GMP 종합보고서 정합성 검토 |
- 종합보고서 초안 - 문서 연결 구조도 - 내부 리뷰 체크리스트 |
✔️ 공정–설비–SOP–기록 간 정합성 ✔️ QA 승인 및 제출 버전 최종화 |
✅ 실무 대응 체크
- 사전상담 시 GMP 운영 상태에 대한
미리 점검된 사실 기반 자료를 제시하고, 전략 수립과 연계해야 합니다 - 혁신의약품 해당 제품의 경우,
GIFT 조건 충족 여부에 대한 명확한 기준 정리가 필요합니다 - 병렬 진행 중 하나의 이탈이나 부실 문서가 전체 지연으로 이어지므로,
사전단계에서 '완결성 확보'가 중요합니다
3️⃣ 공급망 책임 확장 – 공급자 적격성 관리 체계
- 공급망 관리의 중요성:
- 최근 GMP 심사는 제조소뿐만 아니라, 원료의약품(API), 완제 위탁처,
포장·시험 외주 업체 등 외부 공급자의 GMP 이력과 문서 관리 실태까지 확인합니다.
- 최근 GMP 심사는 제조소뿐만 아니라, 원료의약품(API), 완제 위탁처,
- 인증서 제출 시 유의사항:
- WHO나 PIC/S 회원국 발행 인증서 제출 시 실사가 면제될 수 있습니다.
- 하지만 이는 자동 승인이 아닌 문서의 신뢰성과 정합성 평가를 전제로 합니다.
- 기업의 필수 관리 시스템:
- 따라서 기업은 공급자 선정 기준, 재평가 주기, 외주 평가 결과 등을
체계적으로 관리하는 시스템을 갖추어야 합니다.
- 따라서 기업은 공급자 선정 기준, 재평가 주기, 외주 평가 결과 등을
🔸 공급자 평가 흐름도

🔸 공급망 GMP 책임 분담 구조도
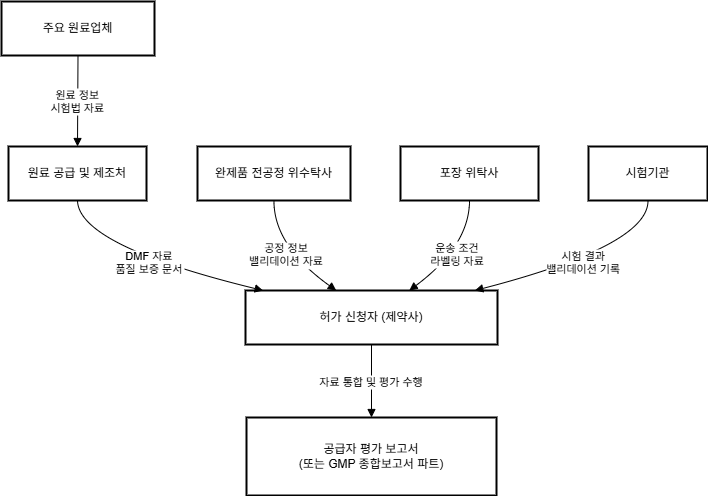
✅ 실무 대응 체크
- 단순 계약서와 COA 제출만으로는 적격성 증빙이 부족하며,
자체 감사결과나 제3자 감사보고서가 필요합니다 - 공급자 재평가 시스템 문서화(SOP, 평가계획서, 평가기록지 등)는 실사 시 점검 대상입니다
- 수입품 공급자 정보는 사전 등록과 추적 가능성 확보가 필요합니다
4️⃣ 간소화된 GMP 자료 – 줄어든 항목, 높아진 내용 깊이
- 제출 자료의 변화:
- 이번 평가 체계 개편으로 서면 제출 자료의 양은 줄어들었으나,
이는 평가 강도가 낮아졌음을 의미하지 않습니다.
- 이번 평가 체계 개편으로 서면 제출 자료의 양은 줄어들었으나,
- 종합보고서의 핵심 요구사항:
- 오히려 종합보고서에는 과거 분산되어 있던
기록, 절차, 평가 결과가 하나의 서술 구조 내에서 유기적으로 정리되어야 합니다. - 특정 항목 누락이나 설명 부족은 문서 전체의 신뢰도를 저하시킬 수 있습니다.
- 오히려 종합보고서에는 과거 분산되어 있던
- 심층적인 요구사항:
- 예를 들어, CQA 설정 기준, 밸리데이션 수행 전략, 일탈 관리 시스템 등의
기술 수준은 과거보다 심층적으로 요구됩니다. - 또한 품질보증 체계에 대한 철학적인 설명도 요구됩니다.
- 예를 들어, CQA 설정 기준, 밸리데이션 수행 전략, 일탈 관리 시스템 등의
✅ 실무 대응 체크
- 평가 자료의 항목 수 감소만으로 준비 시간 단축을 예상하는 것은 잘못된 판단이며,
항목 간 연결성과 핵심 개념 설명력이 전보다 중요 - 기존 SOP 사본 제출 방식에서 '어떻게 운영되는지'를 요약해 설명하는 능력 중심 평가로 전환
- 핵심 품질지표(KPI), 변경관리 절차, 교육훈련 시스템 등도 문서로 설명되어야 평가 가능
🔹 결론 – 준비는 문서가 아니라 시스템으로 시작됩니다
- 정책 개편의 진화:
- 이번 정책 개편은 단순한 절차 변경이 아닌,
제약사의 내부 품질 시스템과 공급망 통제 체계 전반을 평가 체계에 일치시키는 구조로 진화한 것입니다.
- 이번 정책 개편은 단순한 절차 변경이 아닌,
- 문서 작성자의 역할 변화:
- 문서 작성자는 규정 이행 여부를 나열하는 데 그치지 않고,
실제 공정과 시스템 운영이 어떻게 품질 사고를
예방하고 개선 사이클을 유지하는지 설명할 수 있어야 합니다.
- 문서 작성자는 규정 이행 여부를 나열하는 데 그치지 않고,
- 요구되는 준비 체계:
- 모든 대응은 '자료 준비'가 아닌 '시스템 운영' 중심으로 전환되어야 합니다.
- 실무자 입장에서는
문서 작성 → 시스템 검토 → 조직 점검 → 전략 수립이라는 순환적 준비 체계가 요구됩니다.
📌 다음 포스팅에서는 실제 GIFT 제도 및 평가체계 변화가 제조소 운영 매뉴얼,
품질관리 시스템, 교육자료에 어떻게 반영되어야 하는지에 대해
'Data Integrity 관점 해설'로 이어집니다.
전자기록과 수기기록 간 정합성, 현장 일치성 검증 기준 등을 중심으로 분석할 예정입니다.
📚 주요 용어 정리
| 용어 | 정의 |
| GMP 종합보고서 | 기존의 개별 GMP 자료(조직도, 공정도, SOP 등)를 통합하여 제조소의 품질보증 체계, 설비·인력·문서 시스템을 일관된 논리 구조로 설명하는 문서. 병렬심사 및 위험기반 실사의 주요 참조자료로 활용됨. |
| 병렬심사 (Parallel Review) |
기존의 단계별 순차 심사 방식에서 벗어나, GMP, CMC, 임상 자료를 동시에 검토하여 허가 소요 기간을 단축하는 제도. GIFT 트랙에서 핵심으로 작동함. |
| 사전상담 | 허가 신청 전, 식약처와의 사전 협의를 통해 심사 전략을 수립하고 자료 제출 방향을 정하는 절차. 병렬심사 및 조건부 접수의 핵심 기반 단계임. |
| 공급자 평가 시스템 | 원료, 외주처, 수입사 등 공급자에 대한 적격성 평가, 정기 재평가, 문서 관리 등을 포함하는 GMP 요구사항. 최근 GMP 실사에서는 공급망 통제 수준이 평가에 포함됨. |
| 현장 일치성 | 제출된 GMP 문서와 제조소 현장 운영 상태 간의 정합성. 종합보고서나 사전상담 자료와 실제 운영 체계 간 불일치 시 심사 지연 또는 불이익 가능성이 있음. |
| 적격성 평가 (Qualification Assessment) |
자재 또는 위탁 제조소가 GMP 기준을 충족하는지에 대해 실시하는 정량적·정성적 평가. 문서 기반 증거 및 실사 기반 판단 모두 포함. |
| 위험기반 평가 (Risk-Based Evaluation) |
품목·제조소의 위험도에 따라 평가 주기, 심사 강도, 실사 여부를 탄력적으로 조정하는 GMP 심사 원칙. 정기 GMP 평가제도에서 적용됨. |
📕 참고문헌
📌 식품의약품안전처 공식 자료
- 의약품 정책설명회 발표자료 (2025년)
- 주요 제도 개편 요지: GIFT 제도 도입, 병렬심사 체계 전환,
GMP 종합보고서 도입, 시판 전 GMP보고제 폐지, 허가총괄과 신설 등
- 주요 제도 개편 요지: GIFT 제도 도입, 병렬심사 체계 전환,
- 식약처 고시 제2023-87호
- 명칭: 「의약품 제조 및 품질관리 기준」
- 핵심 내용: GMP 기준 및 적합판정서 유효기간 명시 (기본 3년), 변경사항 반영 시 연장 조건 등 제시
- 식약처 민원인 안내서 – GMP 평가 및 실사 민원 안내서
- 주요 항목: GMP 종합보고서 구성, 사전제출자료, 실사 대응 준비 방법 등 실무 대응자료
- 식약처 보도자료 – 규제혁신 3.0 시리즈 (2024년 11월)
- 핵심: 병렬심사, GIFT 트랙의 도입 배경과 행정 효율화 목표 설명
📌 국제 기준 및 가이드라인
- PIC/S PE 009-17 GMP Guide
- 적용범위: 인간용 의약품 제조에 적용되는 국제 GMP 기준
- 내용: 품질시스템(QMS), 밸리데이션, 문서관리, 위생·교육 등 총체적 가이드라인 구성
- ICH Q7~Q12 시리즈 (QbD 및 라이프사이클 관리 중심)
- 주요 지침: Q8(R2)(설계기반 품질), Q9(위험기반 품질관리),
Q10(QMS 구축), Q12(변경관리 라이프사이클 접근 등)
- 주요 지침: Q8(R2)(설계기반 품질), Q9(위험기반 품질관리),
📌 기타 참조자료
- 대한약전 제13개정
- GMP 허가자료 기준과 연계되는 제형 정의 및 공정 특성 기준 제공
- GMP 종합보고서 작성 가이드라인 (KPTA, 자율 기준)
- 실무 적용 시 작성 예시, 각 파트별 필수 항목 및 품질 문서 표준안 제공
반응형
'정책설명회(식약처) > 2025년 의약품 허가 심사' 카테고리의 다른 글
| 06. 글로벌 혁신제품 신속심사(GIFT) 운영 현황 및 계획 (2) | 2025.06.07 |
|---|---|
| 05. 글로벌 혁신제품 신속심사(GIFT) 운영 현황 및 계획 (1) | 2025.06.06 |
| 03. 글로벌 혁신제품 신속심사(GIFT) 운영 현황 및 계획 (1) | 2025.06.04 |
| 02. 글로벌 혁신제품 신속심사(GIFT) 운영 현황 및 계획 (0) | 2025.06.03 |
| 01. 글로벌 혁신제품 신속심사(GIFT) 운영 현황 및 계획 (0) | 2025.06.02 |