
변경관리와 일탈관리:
GMP 품질 시스템의 양축
변경은 통제의 대상, 일탈은 관리의 대상입니다.
두 시스템 모두 GMP의 품질 일관성과 신뢰성 확보에 필수적인 핵심 구성입니다.
🔍 GMP 운영 중 반드시 관리해야 할 두 가지 변화
GMP 환경에서의 모든 변화는 두 가지 주요 유형으로 구분됩니다:
- 계획된 변화는 변경관리(Change Control) 시스템으로 처리합니다.
이는 사전 평가와 승인을 반드시 거쳐야 합니다. - 예상치 못한 상황은 일탈관리(Deviation Management)로 처리합니다.
즉시 보고하고 체계적으로 후속 조치를 취합니다.
이 구분은 단순한 문서 작업이 아닌 품질 보증의 핵심 기반이며
의약품 품질 시스템의 신뢰성을 보장하는 필수 요소입니다.

🧭 개념 정리 – 변경과 일탈은 어떻게 다른가?
GMP 시스템에서 변경과 일탈은 품질관리의 서로 다른 두 도구입니다.
변경관리는 계획된 변화를 미리 검토하고 승인하는 절차이고,
일탈관리는 예상치 못한 문제 발생 시 대응하는 체계입니다.
변경관리는 사전 예방에 중점을 두며,
일탈관리는 문제 해결과 재발 방지에 집중합니다.
두 시스템 모두 의약품 제조의 품질 일관성을 유지하는 데 필수적입니다.
아래 표는 두 시스템의 주요 차이점을 보여주어
실무자들이 상황에 맞는 적절한 관리 방법을 선택할 수 있게 합니다.
| 구분 | 변경관리 | 일탈관리 |
| 정의 | 품질에 영향을 줄 수 있는 사전 변경을 평가하고 승인하는 절차 |
절차, 기준서 등에서 벗어난 상황을 인지하고 관리하는 체계 |
| 목적 | 예방적 품질 통제 | 원인 규명, 재발 방지 중심의 시정조치 |
| 실행 시점 | 변경 전 | 문제 발생 후 |
| 주요 문서 | 변경요청서, 영향평가서등 |
일탈보고서, CAPA보고서등 |
📌 변경 = 사전 예방 / 일탈 = 사후 대응
🧪 실무에서 자주 보는 사례들
GMP 환경에서 변경관리와 일탈관리의 개념적 차이를 이해했다면,
이제 실제 현장에서 어떤 상황이 각 카테고리에 해당하는지
구체적인 사례를 통해 살펴보겠습니다.
아래 표는 제약 산업에서 흔히 발생하는 상황들과 그 분류 근거를 정리한 것입니다.
| 사례 | 분류 | 이유 |
| 제조설비 교체 | 변경관리 | 밸리데이션 및 품질영향평가 필요 |
| 시험기기 신규 도입 | 변경관리 | CSV 적용 여부 평가 대상 |
| 시험기록 누락 | 일탈관리 | 데이터 무결성 위반으로 Root Cause 분석 필수 |
| 라벨 오기 | 일탈관리 | 제품 혼입 위험 →CAPA수립 필요 |
🔄 변경관리 절차: 계획된 변화, 어떻게 통제할까?
변경관리는 체계적인 단계별 접근이 필수적이며,
각 단계마다 철저한 검토와 문서화가 요구됩니다.
아래 6단계 프로세스는 GMP 규정을 준수하는 효과적인 변경관리의 핵심입니다.
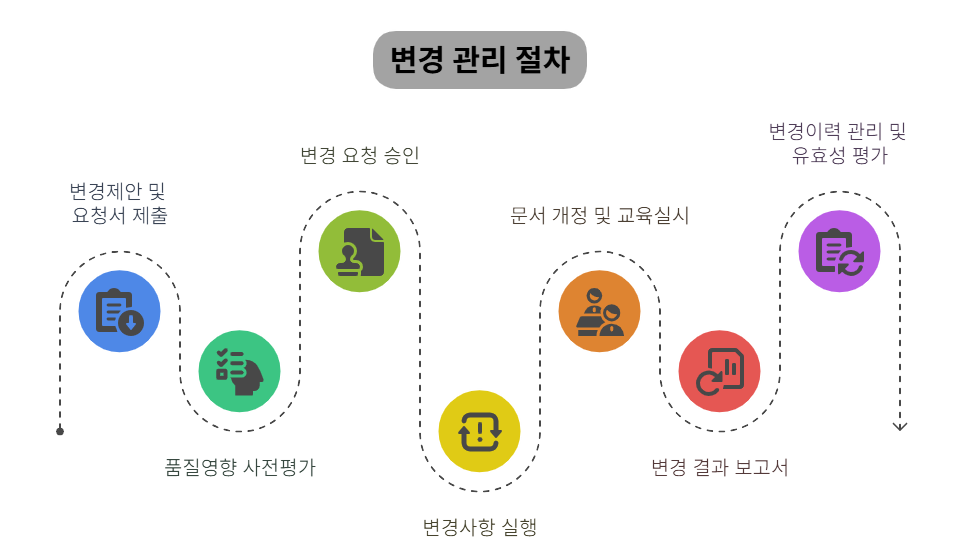
- 변경요청 접수 - 모든 변경의 공식적 시작점
- 변경 분류 (중요/비중요) - 리스크 수준에 따른 관리 전략 결정
- 품질영향평가 - 제품 품질과 규제 요구사항 충족 여부 분석
- 부서별 협의 및 QA 승인 - 다각적 검토와 최종 품질 판단
- 필요 시 밸리데이션, 교육 등 후속 조치 - 변경 검증 단계
- 변경 이력 및 보고서 작성 - 추적성 확보
💡 품목허가 영향 시, 반드시 RA 협의 및 허가변경 병행 → 규제 컴플라이언스 유지
⚠️ 일탈관리 절차: 예기치 못한 상황, 어떻게 대응할까?
일탈이 발생했을 때 신속하고 체계적인 대응은
제품 품질과 환자 안전을 보장하는 핵심입니다.
아래 6단계 프로세스는 GMP 환경에서
일탈 상황을 효과적으로 관리하기 위한 표준 접근법입니다.
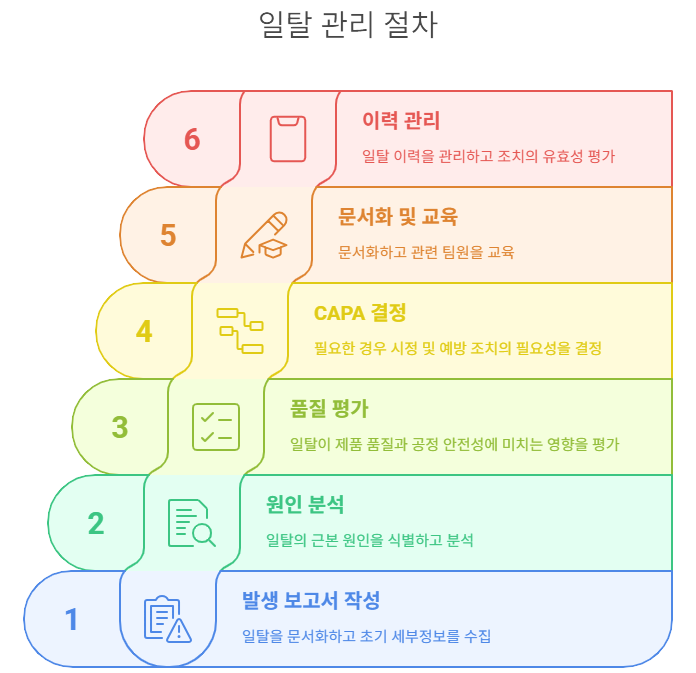
- 일탈보고서 접수 및 작성 - 즉각적 기록과 품질 담당자 통보
- 원인 분석 (RCA) - 근본 원인 규명을 위한 체계적 접근
- 품질영향 평가 - 제품 품질과 환자 안전에 미치는 영향 판단
- 시정·예방조치 수립 (CAPA) - 재발 방지를 위한 실행 계획
- 교육 및 문서 개정 여부 검토 - 조직 학습과 시스템 개선
- 일탈 이력 통합 관리 및 검토 - 트렌드 분석과 패턴 식별
✅ 사소한 일탈도 반드시 문서화하고, 필요 시 CAPA까지 이어져야 함 → 품질문화 구축의 기반
🧩 변경 vs 일탈 – 차이점 요약
아래 표는 변경관리와 일탈관리의 주요 차이점을 정리했습니다.
이 표를 통해 두 시스템의 기본 특성과 운영 방식을 쉽게 확인할 수 있습니다.
이러한 비교는 GMP 품질 시스템 운영에 필수적이며,
규제 기관 점검과 내부 감사 준비에도 중요합니다.
또한 신입 직원 교육과 부서 간 소통에서
두 시스템의 다른 접근법을 이해하는 데 도움이 됩니다.
| 항목 | 변경관리 | 일탈관리 |
| 시점 | 실행 전(사전 통제) | 실행 후(사후 대응) |
| 방향성 | 예방 중심(Preventive) | 시정 중심(Corrective) |
| 핵심문서 | 변경요청서,영향평가서 | 일탈보고서,RCA,CAPA |
| 주요 주체 | QA + 관련부서(협업) | QA 중심, 필요시QC/RA 협업 |
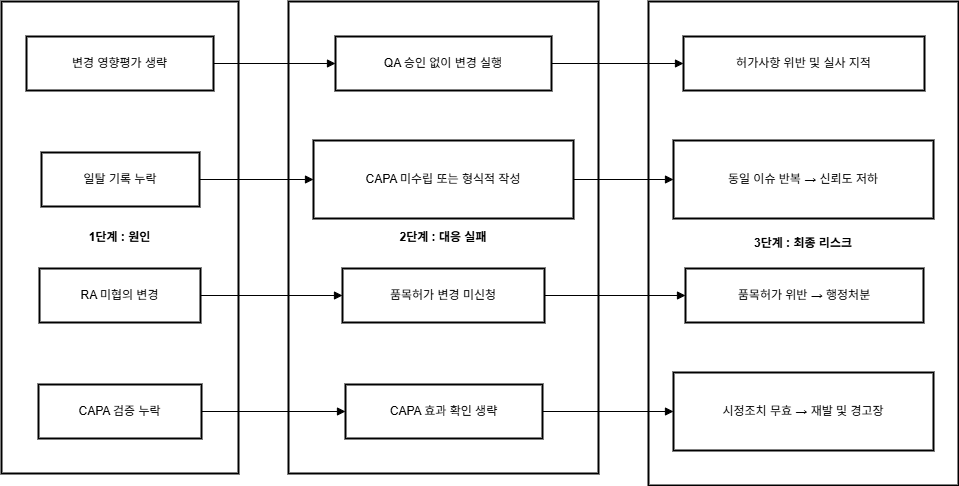
🛠️ 실사에서 자주 지적되는 항목과 대응 방안
변경관리와 일탈관리 시스템은 GMP 실사에서 규제기관이
집중적으로 검토하는 핵심 품질관리 영역입니다.
두 시스템은 의약품 제조의 일관성과 안전성을
보장하는 필수 장치로, 엄격한 관리가 필요합니다.
아래 표는 GMP 실사에서 자주 발견되는 주요 지적사항,
원인, 그리고 개선 방안을 정리했습니다.
이러한 문제점을 미리 파악하고 대응함으로써 규제 준수를
강화하고 품질 시스템의 안정성을 확보할 수 있습니다.
| 지적사례 | 원인 | 개선 포인트 |
| 영향평가 누락 | 변경관리 사전검토미비 | 영향평가 템플릿도입 |
| 보고서 불일치 | 실행과기록불일치 | 실행 후 보고서 작성체계화 |
| 일탈 CAPA 미흡 | 반복적 오류, CAPA형식적수행 | CAPA 효과성 평가포함 |
| 비공식 처리 | 구두 보고,서면 기록누락 | 일탈 보고 기준명확화 및전 직원 교육 |
| 변경→허가변경 누락 | RA 협의누락 | 허가 영향 체크리스트활용 |
🏛️ 품질 시스템의 두 기둥: 핵심 요약
변경관리와 일탈관리는 GMP 품질 시스템의 상호 보완적 관계입니다.
변경은 통제의 대상이고 일탈은 관리의 대상이지만,
두 시스템 모두 의약품 제조의 품질 일관성 유지라는 공통 목표를 가지고 있습니다.
사소한 일탈도 반드시 문서화하고 필요 시 CAPA까지 이어져야 합니다.
품목허가에 영향을 주는 변경은 반드시 RA 협의와 허가변경을 병행해야 합니다.
이러한 철저한 관리를 통해 품질문화 구축의 기반을 마련하고,
규제 컴플라이언스를 유지할 수 있습니다.
궁극적으로 두 시스템의 효과적인 운영은 환자 안전과
제품 품질 보장이라는 GMP의 근본 목적을 달성하는 핵심 수단입니다.
❓ GMP 실무자를 위한 변경·일탈관리 FAQ
Q1. 변경관리와 일탈관리에 ICH Q9의 리스크 기반 접근을 어떻게 적용하나요?
답변:
ICH Q9의 품질 리스크 관리(QRM)를 변경관리(Change Control) 및
일탈관리(Deviation Management)에 통합하면 다음과 같은 이점이 있습니다:
- 리스크 평가 기반 우선순위 설정: 변경 전 영향 평가(FMEA 등)와 일탈의 중요도 분류로 조사·조치 효율 향상
- 리스크 통제: 고위험 변경은 밸리데이션·CAPA 우선, 일탈은 근본원인 분석 후 긴급 CAPA 적용
- 리스크 커뮤니케이션: QRM 결과를 내부 감사 및 PQR에 공유해 지속적 개선 기반 마련
이러한 QRM 통합을 통해 문서화 부담이 감소하고,
중요 항목에 리소스를 집중하여 품질 보증과 규제 대응을 동시에 강화할 수 있습니다.
📚 대표 근거
- ICH Q9(R1) Quality Risk Management, EMA 2023
- WHO QRM & Deviation Management Guidance, WHO 2013
- EMA Annex 11 Change Control 관련 Q&A
Q2. 전산화된 변경관리/일탈관리 시스템에서 Annex 11과 Part 11의 핵심 요건은?
답변:
전자 시스템 기반 변경·일탈관리에서는
EU GMP Annex 11과 FDA 21 CFR Part 11이 핵심 기준입니다:
| 항목 | EU GMP Annex 11 | FDA 21 CFR Part 11 |
| 변경기록 | 변경 요청·영향평가· 승인 절차 문서화 |
Audit Trail로 전자기록 변경사항 자동 기록 |
| 접근 통제 | 최소 권한, 관리자 권한 분리 |
고유 사용자 ID, 인증 절차, 전자서명 확인 |
| Audit Trail | 모든 기록의 변경 이력 추적 가능 |
생성·수정·삭제 시 사용자·시간 기록 |
| 전자서명 | 명확한 규정 없음 (Part 11 준용 필요) |
서명-기록 연계, 이유·시간 포함 필수 |
| 시스템 밸리데이션 | Annex 15 연계 IQ/OQ/PQ 요구 |
변경 시 재검증 포함한 문서화 검증 요구 |
준수 전략:
- 위험 기반 검증: QRM을 적용해 핵심 기능 중심으로 검증
- 통합 QMS 도입: ERP 또는 전용 QMS 모듈 활용
- 모니터링 체계화: 감사로그, CAPA 연계 관리
- 사용자 교육: 전자서명 및 기록 작성 교육 포함
📚 대표 근거
- EMA Annex 11
- FDA 21 CFR Part 11 가이드라인
- GMP Compliance on Computerised Systems
'GMP 기초 > GMP 문서, 규정, 시스템 이해하기' 카테고리의 다른 글
| [GMP 문서, 규정, 시스템 이해하기_05] GMP QMS의 기본 개념: 품질경영시스템의 구조와 실무 적용 (0) | 2025.05.16 |
|---|---|
| [GMP 문서, 규정, 시스템 이해하기_04] GMP 데이터 완전성 개요: ALCOA+ 원칙과 실무적 중요성 (0) | 2025.05.14 |
| [GMP 문서, 규정, 시스템 이해하기_03] GMP 밸리데이션: 품질 일관성과 신뢰성 확보의 과학적 기반 (0) | 2025.05.14 |
| [GMP 문서, 규정, 시스템 이해하기_02] GMP 문서화와 기록관리: SOP, 기록서, 로그북의 정확한 이해 (0) | 2025.05.13 |
| [GMP 문서, 규정, 시스템 이해하기_01] GMP 법령과 가이던스 체계: 실무자가 꼭 알아야 할 규제의 위계 (0) | 2025.05.13 |